
Performance of polygenic risk scores for cancer prediction in a racially diverse academic biobank: Methods part 2
Published:
This blog post is Part 2 of outlining the materials and methods used in our lab’s most recent work on generating polygenic risk scores (PRSs). This article is entitled “Performance of polygenic risk scores for cancer prediction in a racially diverse academic biobank”. It was published in October 2021 and led by Dr. Louise Wang and Heena Desai.
My sources for this blog post are the article, available here on Elsevier and here on PubMed, as well as a text file from Heena that explained the study process in greater detail.
It’s worth noting that the following instructions were created for the purposes of passing on our labs methods to another lab and application in different datasets. In my future work, I actually manipulate this process quite a lot and simplify it. In retrospect, it also has quite a bit of missing information.
Code that manipulates exome data
Below, we use PLINK 1.9 to start manipulating exome data Data used is the Exome .vcf.gz files, of which there is 1 for each chromosome
1. Convert exome.vcf.gz to .bed files for each chromosome
plink –vcf exome_chr#.vcf.gz –make-bed –out chr#_plink_exome
2. Use PLINK to convert pull out only SNPs of interest
- For -snp, enter format of SNPs in PMBB dataset, such as chr#_pos_ref_alt or chr#_pos
- This will create a file for each chromosome, which has subsetted SNPs and still contains the entire population
plink –bfile chr#_plink_exome –snp #_pos_ref_alt –out chr#_pos_genotype –recode A
3. Convert the file master SNP input file into specific columns with headers of “ID” and “rsid”
- These correspond to PMBB ids and whether a given variant at a specific rsid is present for an individual’s genotype with values 0, 1, or 2
cut -d “ “ -f2-2,7- –output-delimiter=, chr#_pos_genotype.raw > chr#_pos_geno.csv
Code that manipulates phenotypic data:
1. Separate individuals into African vs. European ancestry. This was performed using PCA with Eigenstrat fastpca
- Code is not available for this at the moment
- This comes in a .csv filetype, for which I presume there’s now patient IDs in one column and ancestry in another column
3. We then take the .csv file and read it into R
population<-read.csv(pca_file from above, header=T, sep=””)
4. List number of individuals in each ancestry groups
table(population$GIA)
5. Take european population only and save as var
- Write the European subset into a file
- No row names
- Remove quotes
- Tab separated
- Label as “EUR_ancestry_PCs.txt”
EUR_pop <-subset(population, population$GIA==”EUR”)
write.table(EUR_pop, “EUR_ancestry_PCs.txt”, row.names=F, quote=F, sep=”\t”)
6. Extract European subset PMBB IDs and write them to a file
- No row or column names
- Remove quotes
- Tab separated
- Label as “EUR_ancestry_IDs.txt”
EUR_pop_id<-EUR_pop[,c(1,1)]
write.table(EUR_pop_id, "EUR_ancestry_IDs.txt", row.names=F, col.names=F, quote=F, sep="\t")
7. Repeat the same as #5 and #6 but for the African population
AFR_pop<-subset(population, pop$GIA=="AFR")
write.table(AFR_pop, "AFR_ancestry_PCs.txt", row.names=F, quote=F, sep="\t")
AFR_pop_id<-AFR_pop[,c(1,2)]
write.table(AFR_pop_id "AFR_ancestry_IDs.txt", row.names=F, col.names=F, quote=F, sep="\t")
Code that manipulates genetic data
8. Keep European individuals, and remove related individuals
- We have a “EUR_ancestry_IDs.txt” file
- We have a “related individuals file” with PMBB ids
- For each chromosome with a filetype of .vcf.gz
- Keep the european individuals
- Remove the related individuals
- Save the remaining files as .bed format
plink --vcf VCF_chr#.vcf.gz --keep EUR_ancestry_IDs.txt --remove (related individuals file) --make-bed --out EUR_file_name_chr#
9. Merge the list of 23 chromosome files into one .txt file, then convert each to .bed filetype
ls EUR_file_name_chr*.bed > eur_merge_list.txt
plink --merge-list eur_merge_list.txt --allow-no-sex --make-bed --out EUR_file_name_chrALL
10. Remove samples that failed sex check
plink –bfile –remove failed_sex_check_file –make-bed –out EUR_file_name_final_chrALL
11. Repeat steps 8-10, but for African population
plink --vcf VCF_chr#.vcf.gz --keep AFR_ancestry_IDs.txt --remove (related individuals file) --make-bed --out AFR_file_name_chr#
ls AFR_file_name_chr*.bed > merge_list.txt
plink --merge-list afr_merge_list.txt --allow-no-sex --make-bed --out AFR_file_name_chrALL
plink --bfile AFR_file_name_chrALL --remove failed_sex_check_file --make-bed --out AFR_file_name_final_chrALL
End result: we now have two files of .bed format:
- EUR_file_name_final_chrALL
- AFR_file_name_final_chrALL
Phenotyping of PMBB participants
I explain the phenotyping of PMBB participants in greater detail in prior entries. Briefly, at the time of the study, there were 19,935 individuals included for whom electronic health data was available. This includes demographic data, lab results, and ICD-9 and ICD-10 diagnosis codes.
Interestingly, the presence of an ICD code for prostate cancer was imperfect. Manual chart review was performed for 2365 individuals with at least 1 ICD-9/10 billing code for prostate cancer. The positive predictive value of 1 ICD-9/10 prostate cancer billing code was 94%, while the presence of >5 ICD-9/10 prostate cancer billing codes for a patient across several encounters had much high positive predictive value.
Because of this finding, manual chart review was performed for several other cancers which showed slightly better positive predictive value of ICD-9/10 codes for true presence of a cancer diagnosis.
SNV (Single Nucleotide Variation) selection
Summary statistics were obtained using the most recent study and largest GWAS available in the GWAS catalog for each of the 15 cancers in this study. Most of these were obtained at the www.ebi.ac.uk website, and the table below shows where they were taken from.
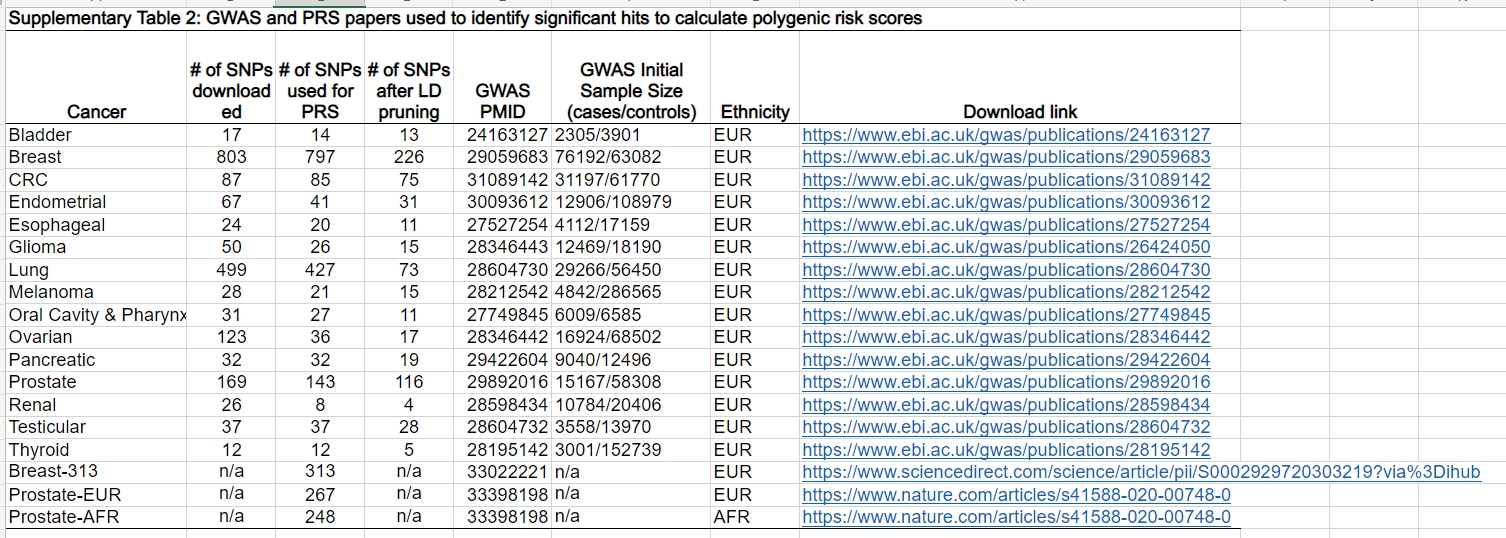
These were studies of European populations. SNVs were chosen on the basis of a P value threshold of P < 1 × 10–6.
SNVs were also controlled for linkage disequilibrium (LD). If there was a pairwise genotypic correlation at r2 > 0.1, then SNVs were “pruned”. I.e, only the most significant SNV in that group was included.
PRS generation and statistical analysis
PRS for each individual was calculated using PLINK 1.9 by summing the LD-pruned SNV variants and weighing their corresponding effect sizes using the odds ratios (ORs) or beta values reported in the original GWAS. We standardized each cancer PRS to a mean of 0 and SD of 1 and used logistic regression to test for the association between cancer PRS and cancer phenotype and to compare the top versus bottom quintiles of polygenic risk, controlling for age, sex, and the first 10 within-ancestry PCs as covariates. We performed a primary analysis for cancers with >100 cases and included a secondary analysis of the remaining cancer types. Area under the curve (AUC) for primary phenotype was determined using the package pROC.23 All statistical analyses were performed using R 4.0.3.
Code for generation of PRSs
1. Select out only variants for our SNPs that achieved genome wide significance in GWAS studies
- This filetype is named gws_variants_file.txt
Here, we’re inputting our final .bed file that was created above, for each our African and European population
- EUR_file_name_final_chrALL
- AFR_file_name_final_chrALL
Our output file is a .profile file that should include
- A master file of genome-wide significant SNPs for each individual
- Columns that identify covariant IDs, allele codes, and allele scores
- Note that 3 4 7 here denotes columns for: variant IDs - col 3, allele codes - col 4, scores - col 7
plink --bfile EUR_file_name_final_chrALL --score gws_variants_file.txt 3 4 7 header --out EUR_cancer_name_PRS_PLINK
plink --bfile AFR_file_name_chrALL --score gws_variants_file.txt 3 4 7 header --out AFR_cancer_name_PRS_PLINK
2. Load libraries
library(pROC)
library(ggplot2)
library(scales)
library(tidyverse)
library(dplyr)
3. Add our 10 principal components
- Add covariates that were measured through PCA - this is essentially 10 control variables
- Ensure labelling of “id” as our first column throughout all files
covariates <- read.csv(file= eur_covariate_file, header = T, sep="\t")
colnames(eur_covariates)[1] = "id"
4. Idenfity subpopulation with the cancer in question
- Read the individuals into a variable named “phenotypes”
phenotypes <- read.csv("Phenotypes/EUR_cancer_name_Pheno.csv", header =T, sep=",", check.names = F)
colnames(phenotypes)[1] = "id"
colnames(phenotypes)[2] = "pheno"
5. Load .profile file into variable, label first column as “id”
genotype file = .profile generated from PLINK
genotypes <- read.csv("EUR_cancer_name_PRS_PLINK.profile", header = T, sep = "")
colnames(genotypes)[1] <- "id"
6. Scale genotype file
genotypes_scaled <- data.frame(genotypes[2], (apply(genotypes[6],2,scale)))
colnames(genotypes_scaled)[1] = "id"
colnames(genotypes_scaled)[2] <- "normalizedScore"
7. Merge 3 files based on “id” column:
- Genotype file
- Phenotype file
- Covariate file
score_pheno = merge(genotypes_scaled, phenotypes, by="id")
score_pheno_cov = merge(score_pheno, covariates, by="id")
8. Extract top, bottom, and remaining quintiles
top = which(score_pheno_cov$normalizedScore > quantile(score_pheno_cov$normalizedScore, 0.8))
bottom = which(score_pheno_cov$normalizedScore < quantile(score_pheno_cov$normalizedScore,0.2))
rest = which(score_pheno_cov$normalizedScore <= quantile(score_pheno_cov$normalizedScore, 0.8))
9. Define data frames for top, bottom, and remaining quintiles
top_q = score_pheno_cov[top,]
bot_q = score_pheno_cov[bottom,]
top_bot = c(top_q, bot_q)
10. Regression model as below:
mod <- glm(score_pheno_cov$pheno ~ score_pheno_cov$normalizedScore + score_pheno_cov$gender + score_pheno_cov$Age + score_pheno_cov$PC1 + score_pheno_cov$PC2 + score_pheno_cov$PC3 + score_pheno_cov$PC4 + score_pheno_cov$PC5 + score_pheno_cov$PC6 + score_pheno_cov$PC7 + score_pheno_cov$PC8 + score_pheno_cov$PC9 + score_pheno_cov$PC10, family=”binomial”)
summary(mod)
11. Repeat Steps 1-10 for AFR population
That’s all for today!